This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Introduction
Major depression is a severe mental illness that has debilitating effects on an individual’s daily life and is a major contributor to early mortality due to suicide [1]. It is the most commonly diagnosed form of depression and affects about 7% of all adults in the United States [2]. There are a variety of symptoms in major depression but the two major symptoms required to be diagnosed with major depression as described by the American Psychiatric Association are depressed mood or loss of interest or pleasure for at least a period of two weeks.
There are many environmental factors that can cause major depression but these factors interact with our genetic makeup to increase a person's risk of developing major depression. Genes encode for proteins and structures involved in the functioning of our brain such as neurons and neurotransmitters. Hence, people with more vulnerable genetic makeup are more predisposed to developing major depression. First-degree family members of individuals with major depressive disorder have a fourfold higher risk of developing major depression than the general population and monozygotic twin studies have shown that the heritability of major depression is 37% [3. 4].
To understand the biological basis of major depression, we must first understand how development of the brain occurs. The synaptic connections of the brain gets refined as a person grows from childhood to adulthood, resulting in an overall decrease in the number of synaptic connections in adulthood compared to a 2-year old child (Fig.1). This refinement is done via the elimination of inhibitory and excitatory neurons that make up synaptic connections. Imbalance in the number of inhibitory and excitatory neurons (synaptic density) is thought to lead to a higher risk of major depression [5].
There are many environmental factors that can cause major depression but these factors interact with our genetic makeup to increase a person's risk of developing major depression. Genes encode for proteins and structures involved in the functioning of our brain such as neurons and neurotransmitters. Hence, people with more vulnerable genetic makeup are more predisposed to developing major depression. First-degree family members of individuals with major depressive disorder have a fourfold higher risk of developing major depression than the general population and monozygotic twin studies have shown that the heritability of major depression is 37% [3. 4].
To understand the biological basis of major depression, we must first understand how development of the brain occurs. The synaptic connections of the brain gets refined as a person grows from childhood to adulthood, resulting in an overall decrease in the number of synaptic connections in adulthood compared to a 2-year old child (Fig.1). This refinement is done via the elimination of inhibitory and excitatory neurons that make up synaptic connections. Imbalance in the number of inhibitory and excitatory neurons (synaptic density) is thought to lead to a higher risk of major depression [5].
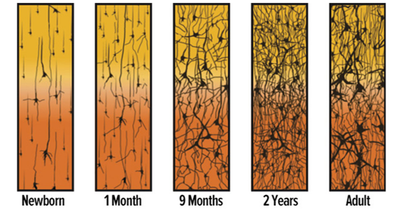
Fig.1: Development of the human brain showing number of synaptic connections throughout different life stages.
The gene associated with major depression is Myocyte Enhancer Factor 2C (MEF2C). Genome wide association studies have shown the presence of single nucleotide polymorphisms (SNPs) near the MEF2C gene locus in individuals of European and Australian descent with depression [6,7]. MEF2C is a transcription factor that regulates synaptic density during the development to the brain [5]. Transcriptional activation of MEF2C is thought to be dependent on phosphorylation by Extracellular-signal-regulated kinase 5 (ERK5) and p38 mitogen-activated protein kinase (p38 MAPK). Protein interaction networks of MEF2C also shows that MEF2C interacting proteins are involved in neuron differentiation, apoptosis and most importantly, the MAPK cascade (Fig.2). Yet, little is known about how phosphorylation of MEF2C affects its role in regulating synaptic density in depression.
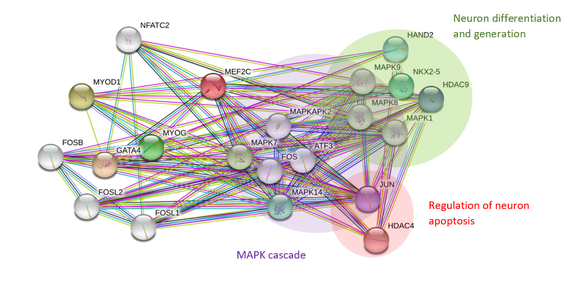
Fig.2: Protein interaction network of MEF2C.
My primary goal is to understand the role of phosphorylation on MEF2C in the regulation of synapse elimination during brain development. My hypothesis is that mutation of conserved phosphorylation sites in MEF2C will result in improper elimination of excitatory neurons, leading to an imbalance in the number of excitatory and inhibitory neurons.
To study the role of MEF2C in depression, I will use zebrafish as my model organism. Zebrafish share major neurotransmitters, hormones, and relevant receptors with mammals, making them a suitable model organism for studying synaptic development [9]. More importantly, zebrafish are also highly social animals, making depressive phenotypes easy to observe. Zebrafish with depressive phenotype show a global reduction of swimming, social withdrawal indicated by a lack of swimming in shoals and a droopy tail angle when at rest (Fig. 3)
To study the role of MEF2C in depression, I will use zebrafish as my model organism. Zebrafish share major neurotransmitters, hormones, and relevant receptors with mammals, making them a suitable model organism for studying synaptic development [9]. More importantly, zebrafish are also highly social animals, making depressive phenotypes easy to observe. Zebrafish with depressive phenotype show a global reduction of swimming, social withdrawal indicated by a lack of swimming in shoals and a droopy tail angle when at rest (Fig. 3)
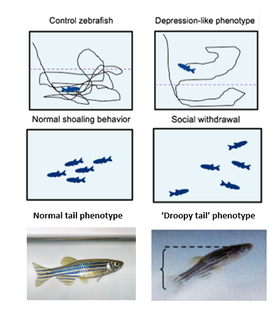
Fig.3: Behavior of depression phenotypes of zebrafish (right) compared to wildtype zebrafish (left).
Aim 1: Identifying conserved phosphorylation sites across MEF2C homologs involved in neuronal development
To identify evolutionarily conserved phosphorylation sites, I will use MEGA and CLUSTAL OMEGA to do sequence alignment of the MEF2C protein in homologs obtained using ENSEMBL. I will then do a screen using zebrafish by mutating these phosphorylation sites using CRISPR/Cas9 and identify mutants with behavioral phenotypes indicative of depression. The brains of individuals with depressive phenotypes will be examined with immunohistochemical markers for excitatory and inhibitory neurons to measure synaptic density. The reason for identifying conserved phosphorylation sites is because the more conserved a protein, the more likely the protein is important in the function of an organism. I hypothesize that zebrafish with mutations in conserved phosphorylation sites will have phenotypes associated with depression and abnormal synaptic density since MEF2C regulates neuronal development. This is because mutations in conserved phosphorylation sites affects transcriptional activation of MEF2C, resulting in abnormal elimination of excitatory synaptic neurons.
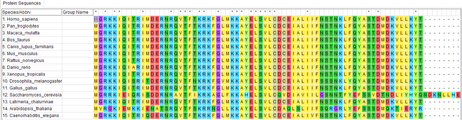
Fig.4: Sequence allignment using MEGA to identify conserved phosphorylation sites.
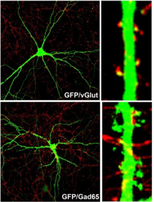
Fig. 5: Neuronal markers (red) used to determine synaptic density. vGlut stains excitatory neurons and Gad65 stains inhibitory neurons.
|
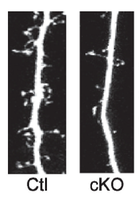
Fig.6: Predicted synaptic densites of MEF2C mutant (right) and wildtype zebrafish (left).
|
Aim 2: Identifying differently expressed genes important in maintaining synaptic density in development
I will isolate mRNA from brain tissue of phosphorylation mutant MEFC2 zebrafish (from Aim1) and wildtype (WT) individuals and conduct RNA-seq. I will then identify differentially expressed genes (DEGs) between mutants with and without depressive phenotypes and classify the functions of these genes using Gene Ontology. I will conduct a loss of function screen with CRISPR/Cas9 using these statistically significant DEGs and examine synaptic density in WT and mutant brains using immunohistochemistry. I predict that zebrafish mutants that display depressive phenotypes have different gene expression profiles for genes involved in neuron development as well as the MAPK cascade compared to mutants without depressive phenotypes and WT individuals. This is because genes involved in neuron development elimination will be differentially expressed as they might not be transcriptionally activated or repressed by mutated MEF2C.
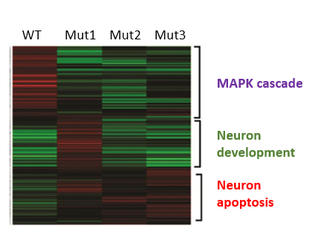
Fig.7: Predicted differences in DEGs in wildtype and mutant zebrafish.
Aim 3: Identifying kinases responsible for MEF2C phosphorylation important in synaptic density
I will conduct pull-down/mass spectrometry using tandem affinity purification (TAP)-tagged baits made from regions of MEF2C containing conserved phosphorylation sites. Mass spectrometry is then used to identify these interacting proteins and Gene Ontology is used to assess the functions and identity of these proteins, specifically looking for kinases that bind to WT but not mutant MEF2C. I will use CRISPR/Cas9 to conduct a loss of function screen for these kinases and measure synaptic density in WT and mutant brains using immunohistochemistry to validate the role of these kinases in synaptic density regulation. Identifying proteins that interact with the regions of MEF2C containing phosphorylation sites will allow us to understand which specific kinases interact with MEF2C to regulate synaptic density. I hypothesize that kinases that bind to WT MEF2C but are unable to bind to mutant MEF2C are important in the phosphorylation of MEF2C in regulating synaptic density.
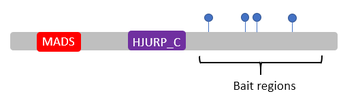
Fig.8: Regions of MEF2C containing phosphorylation sites that were used to make baits.
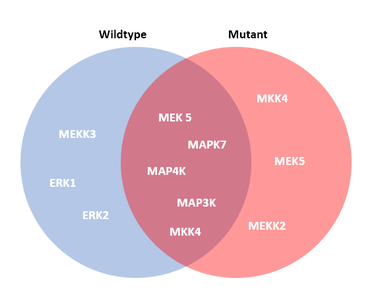
Fig.9: Predicted results of different kinases binding to wildtype and mutant MEF2C. Kinases that bind to only WT but not mutant MEF2C are important in synaptic regulation.
Conclusions & Further Direrctions
Identifying how phosphorylation affects the role of MEF2C in regulating synaptic density balance is important in establishing a biological pathway for depression. This will allow the development of therapeutics aimed at rebalancing synaptic density in patients with depression due to defects in MEF2C phosphorylation.
The next steps in developing therapeutics would be to create drugs targeting the pathways of regulation of MEF2C. This involves targeting kinases or phosphorylation sites important in synaptic density regulation as identified in AIm 3. This would allow the rebalancing of synaptic density to reduce the effects of depression. Another method would be to target the rejuvenation of neurons directly to rebalance synaptic density. Drug screens for depression should be done with drugs that cause neuron regeneration via the regulation of MEF2C.
The next steps in developing therapeutics would be to create drugs targeting the pathways of regulation of MEF2C. This involves targeting kinases or phosphorylation sites important in synaptic density regulation as identified in AIm 3. This would allow the rebalancing of synaptic density to reduce the effects of depression. Another method would be to target the rejuvenation of neurons directly to rebalance synaptic density. Drug screens for depression should be done with drugs that cause neuron regeneration via the regulation of MEF2C.
| jer_final_presentation_4.5.18.pdf |
| jer_final_presentation_5.3.18_.pdf |
References:
[1] Mullins, N., & Lewis, C. M. (2017). Genetics of depression: progress at last. Current psychiatry reports, 19(8), 43.
[2] https://www.nimh.nih.gov/health/statistics/major-depression.shtml
[3] Diagnostic and Statistical Manual of Mental Disorders: https://dsm.psychiatryonline.org/doi/full/10.1176/appi.books.9780890425596.dsm04
[4] Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157(10):1552–62.
[5] Harrington, A. J., Raissi, A., Rajkovich, K., Berto, S., Kumar, J., Molinaro, G., ... & Huber, K. M. (2016). MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife, 5.
[6] Hyde, C. L., Nagle, M. W., Tian, C., Chen, X., Paciga, S. A., Wendland, J. R., ... & Winslow, A. R. (2016). Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nature genetics, 48(9), 1031.
[7] Corfield, E. C., Yang, Y., Martin, N. G., & Nyholt, D. R. (2017). A continuum of genetic liability for minor and major depression. Translational psychiatry, 7(5), e1131.
[8] Wang, Y., Liu, L., & Xia, Z. (2007). Brain‐derived neurotrophic factor stimulates the transcriptional and neuroprotective activity of myocyte‐enhancer factor 2C through an ERK1/2‐RSK2 signaling cascade. Journal of neurochemistry, 102(3), 957-966.
[9] Fonseka, T. M., Wen, X. Y., Foster, J. A., & Kennedy, S. H. (2016). Zebrafish models of major depressive disorders. Journal of neuroscience research, 94(1), 3-14.
Images:
Header: http://www.futureforall.org/gallery/microscope-images-17.html
Fig.1: https://fuzzyscience.wikispaces.com/pruning
Fig.7: https://www.researchgate.net/figure/Heat-map-visualization-and-pathway-analysis-of-differentially-expressed-genes-a_fig2_284797148
[1] Mullins, N., & Lewis, C. M. (2017). Genetics of depression: progress at last. Current psychiatry reports, 19(8), 43.
[2] https://www.nimh.nih.gov/health/statistics/major-depression.shtml
[3] Diagnostic and Statistical Manual of Mental Disorders: https://dsm.psychiatryonline.org/doi/full/10.1176/appi.books.9780890425596.dsm04
[4] Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157(10):1552–62.
[5] Harrington, A. J., Raissi, A., Rajkovich, K., Berto, S., Kumar, J., Molinaro, G., ... & Huber, K. M. (2016). MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife, 5.
[6] Hyde, C. L., Nagle, M. W., Tian, C., Chen, X., Paciga, S. A., Wendland, J. R., ... & Winslow, A. R. (2016). Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nature genetics, 48(9), 1031.
[7] Corfield, E. C., Yang, Y., Martin, N. G., & Nyholt, D. R. (2017). A continuum of genetic liability for minor and major depression. Translational psychiatry, 7(5), e1131.
[8] Wang, Y., Liu, L., & Xia, Z. (2007). Brain‐derived neurotrophic factor stimulates the transcriptional and neuroprotective activity of myocyte‐enhancer factor 2C through an ERK1/2‐RSK2 signaling cascade. Journal of neurochemistry, 102(3), 957-966.
[9] Fonseka, T. M., Wen, X. Y., Foster, J. A., & Kennedy, S. H. (2016). Zebrafish models of major depressive disorders. Journal of neuroscience research, 94(1), 3-14.
Images:
Header: http://www.futureforall.org/gallery/microscope-images-17.html
Fig.1: https://fuzzyscience.wikispaces.com/pruning
Fig.7: https://www.researchgate.net/figure/Heat-map-visualization-and-pathway-analysis-of-differentially-expressed-genes-a_fig2_284797148
